Éste artículo fue publicado en el año 1980 firmado por J.Martín Mira y C.Santos García. Aunque han pasado treintaicinco años, todavía puede ser útil a aquellos investigadores que no tengan noticia de algunos de los datos que se citan.
Con los tres artículos del blog que siguen a éste, se cierra un conjunto temático que quizá sea publicado por otros medios en un próximo futuro.
Y para cerrar esta breve aportación informo, para quien pueda estar interesado, de la publicación reciente de un libro en el que relato mis experiencias personales y profesionales en el terreno científico. El libro se titula «Apuntes tomados a través de una vida» de Juan Martín Mira y Editado por Letras de Autor. En Amazon y la Casa del Libro está en edición digital.
Estructura superficial de las partículas en emulsión O/W y de las dispersiones coloidales de los áridos minerales
Los procedimientos para fabricar una emulsión bituminosa O/W implican la mezcla del betún asfáltico con el medio acuoso. Para lograr el emulsionamiento son precisas dos condiciones: dispersar finamente el betún en el medio y mantener la dispersión. La primera condición se puede conseguir mediante la energía mecánica aportada por molinos coloidales, homogeneizadores, ultrasonidos, etc., obteniéndose partículas esféricas debido a la tensión interfacial. La segunda requiere la estabilización de las partículas bituminosas, que tienden a aglomerarse espontáneamente para reducir el área interfacial, con la consiguiente reducción de energía libre; en la práctica se obtiene con tensioactivos catiónicos y aniónicos, aunque también se emplean macromoléculas de carácter no-iónico.
Harkins (1), en 1952, supuso que la partícula dispersa en emulsión O/W estaba constituida por un núcleo de la sustancia oleosa en la que se adsorbían los radicales hidrofóbicos del tensioactivo, quedando en su superficie los grupos polares hidrofílicos. Se puede establecer que la estructura global de la partícula responde a la estructura micelar descrita por Stigter (2), y que ha sido adaptada a una partícula bituminosa (considerada plana por simplicidad) en emulsión catiónica, como se muestra en la Figura 1. Por ser el medio acuoso una disolución de electrolitos, se crea en la interfase partícula-disolución una estructura de capa doble constituida por una capa rígida, de Stern, y por una capa difusa, de Gouy. La capa doble es la responsable de la estabilidad de la emulsión.
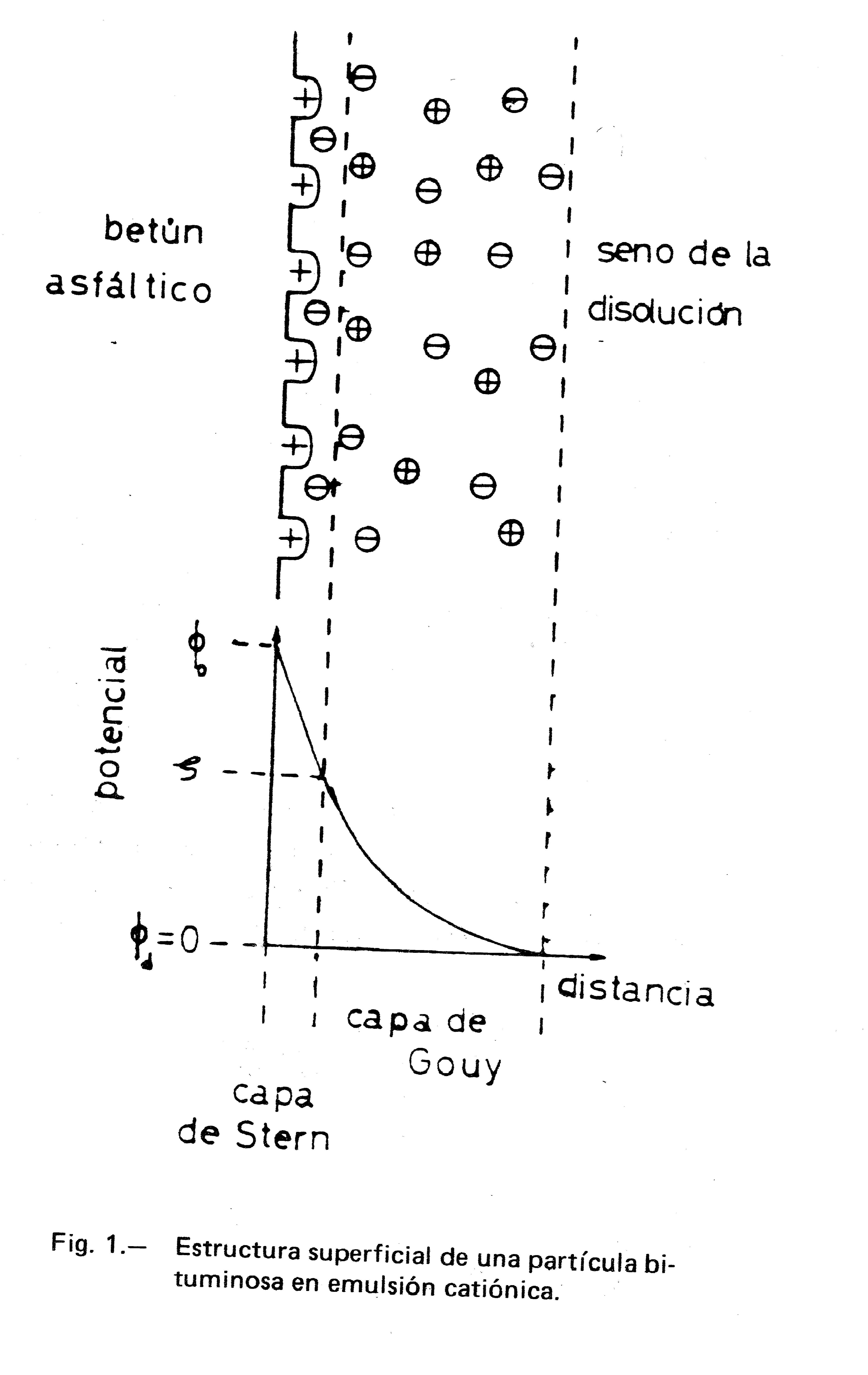
La carga positiva de la capa de Stern se encuentra compensada por un exceso de contraiones situados en la capa de Gouy. En la misma figura se definen los potenciales: el potencial zeta ζ se asimila al potencial de la capa rígida y equivale a la diferencia entre el potencial de dicha capa, φs, y el potencial de la disolución que se toma como nulo.
La estabilidad electrocinética de las partículas en emulsión depende de las fuerzas atractivas y repulsivas entre ellas. Cuando dos partículas se aproximan, habrá una repulsión coulombiana, ya que las dos capas difusas contienen cargas del mismo signo. El alcance de esta repulsión es aproximadamente igual al espesor de la capa difusa.
La estructura de la capa descrita es asignable, con retoques, a la dispersión de un polvo mineral calizo o silíceo en agua o en disoluciones acuosas. La superficie de la partícula coloidal presentará iones de un determinado signo que crean una adsorción de contraiones formando una capa difusa. Consecuentemente, tanto las partículas de las emulsiones bituminosas como las coloidales de un polvo mineral, poseerán un potencial electrocinético medible.
Medida del potencial zeta
Los fenómenos electrocinéticos surgen cuando las fases dispersa y continua de la emulsión o dispersión se mueven relativamente, no permaneciendo estática la capa doble. Son de cuatro clases: potencial de sedimentación, electro-ósmosis, potencial de flujo y electroforesis. En todos ellos se pone de manifiesto una partícula cinética que incluye la capa de Stern donde está la superficie de corte y donde se mide el potencial zeta. El método más empleado para la medida de ζ en emulsiones y dispersiones coloidales es el electroforético, que consiste en el movimiento de la partícula cargada bajo la acción de un campo eléctrico constante. Los dos procedimientos electroforéticos usuales de medida son el del frente móvil y el microelectroforético. Como el primero requiere que el sistema disperso tenga un color diferente al del medio de dispersión, es más frecuente el empleo del segundo. Con los diferentes métodos de medida se pretende evaluar la velocidad electroforética, v, a partir de la cual se calcula el potencial zeta mediante la sencilla relación de Smoluchoswki: ζ= ἠ v / ὲ0 ὲr E , donde ἠ es la viscosidad del medio de dispersión, ὲ0 ὲr su permitividad eléctrica y E el potencial aplicado.
Potencial zeta en dispersiones catiónicas y en áridos silíceos
Riddick (3) ha medido la variación del potencial zeta con la concentración de una sílice coloidal de 1,1 µm de diámetro, separada por flotación de aire del producto de machaqueo de una arenisca. Obtiene los valores preparando mezclas de sílice coloidal en agua destilada en proporciones del 0,4 al 50 por ciento, centrifugando y añadiendo a cien mililitros del líquido que sobrenada una gota de la lechada original, cantidad suficiente para efectuar las medidas electroforéticas. El valor de ζ= – 30 ± 2 mV que resulta para todo el intervalo de concentraciones ensayado indica que la partícula cinética tiene una carga neta negativa.
En la Figura 2, tomada de Li y De Bruyn (4), se denota la variación del potencial zeta con el pH para un cuarzo cristalino machacado y tamizado, al que se lava con HCl 1N en caliente para eliminar el hierro y, posteriormente, con agua destilada. Después de un nuevo machaqueo y de colectar la fracción de 5 – 20 µm, se trata con HF al diez por ciento para eliminar la capa formada en el proceso de trituración, se lava con NaOH 0,1N y con agua de conductividad. Puede verse que el valor del potencial zeta en el intervalo de pH de 3,5 a 7 es más negativo que en el caso de la sílice coloidal.

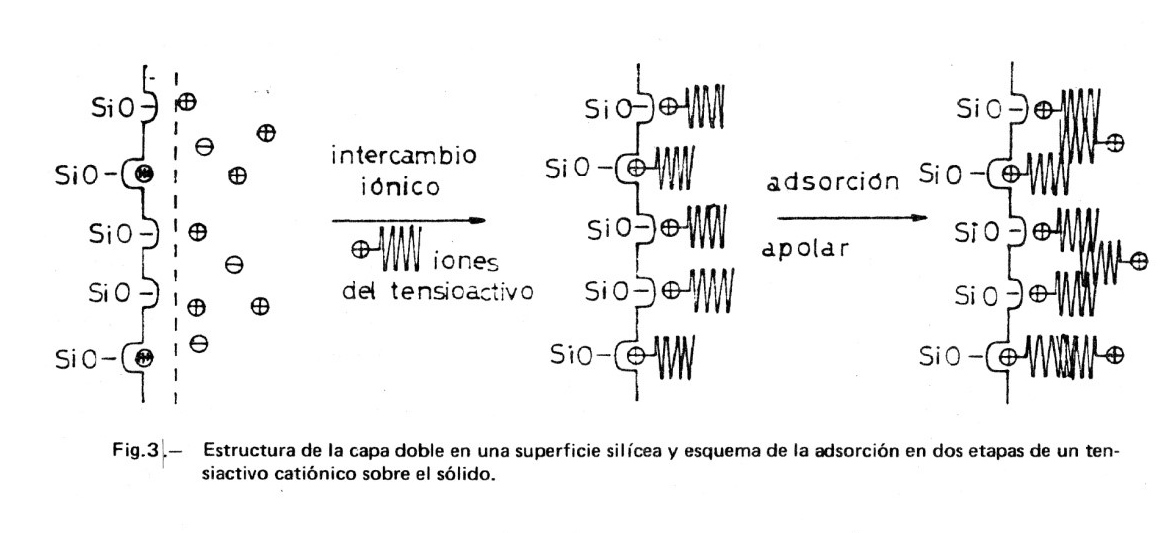
En la Figura 3 se muestra una posible estructura de la capa doble en una superficie silícea en agua pura. Corresponde al modelo sugerido por Dobiá (5) en el que se han introducido los retoques necesarios para mostrar la estructura de la capa doble. El número de H+ en la partícula cinética depende del grado de disociación, el cual depende a su vez de la acidez o basicidad del medio. La carga de la capa de Gouy será tal que compense la carga de la capa de Stern. Si existen además otros tipos de iones en el medio, se repartirán entre las capas, según su capacidad de adsorción e intercambio. En medio ácido se reprime la disociación de los grupos silanol, SiOH, habiendo más H+ adsorbidos en la capa de Stern: la partícula cinética presenta una carga menor y, en consecuencia, el valor de ζ se aproxima a cero. En medio básico, aumentará la carga negativa de la superficie por hacerse mayor el grado de disociación.
En la misma Figura 3 se esquematiza el modelo simplificado de Dobiá para la adsorción de iones de iones de tensioactivo catiónico sobre la superficie silícea polar. Pueden suponerse dos etapas: a) Una adsorción polar en la primera capa, dándose un intercambio iónico entre los H+ (u otros cationes inorgánicos adsorbidos) y los cationes del tensioactivo, que se completa con un apareamiento iónico entre las capas negativas de la superficie y las positivas del adsorbato; la capa completa tiene carácter apolar y el potencial electrocinético será nulo. b) Una adsorción apolar del tensioactivo sobre la primera capa adsorbida, en virtud de las fuerzas de van der Waals entre los radicales hidrocarbonados del tensioactivo; la capa adquiere carácter polar, estando su carga compensada por iones negativos (contraiones) situados en la capa de Stern y en la capa de Gouy.
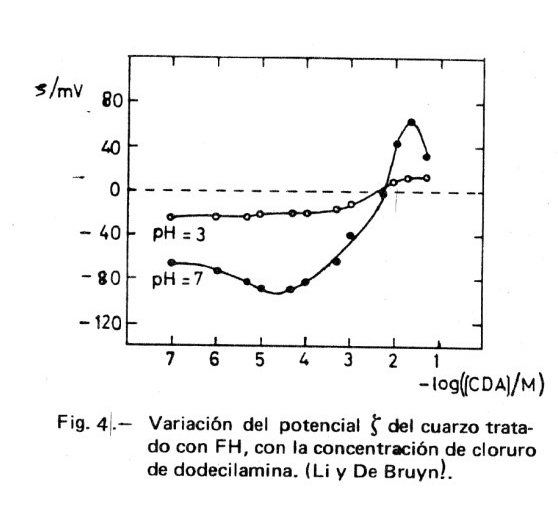
El mecanismo descrito está en acuerdo cualitativo con los valores dados por Li y De Bruyn (4) que se recogen en la Figura 4. Al aumentar la concentración del tensioactivo CH3-(CH2)10-CH2-NH3+.Cl– (clorhidrato de dodecilamina, CDA), se adsorbe sobre el cuarzo hasta formar una primera capa que conduce a un valor de ζ= 0; la posterior adsorción de una segunda capa de DA+ lleva a valores de ζ positivos. El hecho de que se alcance un punto en el que la carga de la partícula cinética sea nula ( ζ = 0), no quiere decir que la carga superficial sea cero, sino que la carga de la capa de Stern ha alcanzado la electroneutralidad.
Cuando la superficie sobre la que se adsorben los iones del tensioactivo es diferente a la sílice, las cosas suceden de manera análoga a la expuesta. Cabe decir que, en general, los sistemas coloidales naturales presentan un potencial zeta negativo. En la Figura 5, compuesta con datos de diferentes autores para la adsorción de un mismo tensioactivo, bromuro de dodeciltrimetilamonio (BDTA), CH3-(CH2)10-CH2-NH(CH3)3+.Br–, puede verse que el potencial electrocinético nulo no se alcanza con la misma concentración de tensioactivo, ni son tampoco iguales las magnitudes de carga positiva que adquiere la superficie. La curva (a) de la Figura 5 corresponde a un sol de yoduro de plata (6) con un pI de valor 4 conseguido añadiendo yoduro de potasio; en la curva (b) se mide la adsorción del BDTA en un látex de poliestireno monodisperso de 0,761 ± 0,022 µm con grupos iónicos carboxilo que ocupan un área de 5,44 nm2 por grupo cargado (7); la curva (d) corresponde a un látex de polimetilmetacrilato de 0,22 µm de diámetro de partícula (8): en los tres sistemas (a, b, d) el punto de carga cero se establece cuando la concentración de BDTA se sitúa en el entorno de 10-5 mol/L. En el caso, descrito en la curva (c), de una sílice coloidal precipitada y a pH = 8 (9) no se consigue el punto de carga cero hasta que la concentración es de, aproximadamente, 3.10-3 mol/L. Lane y Ottewill (10) miden la variación del potencial zeta con la concentración de BDTA para una emulsión de betún asfáltico, representada en la curva (e) de la Figura 5, un sistema en el que no se puede obtener un valor del punto de carga cero. No obstante, en la figura puede verse que su comportamiento es parecido al del látex.
Los datos acumulados en la Figura 5 denotan las características diversas con respecto al intercambio iónico y a la interacción electrostática de las superficies descritas con el ion DTA+. En la zona de potencial zeta positiva, la estructura superficial de las partículas dispersas puede ser representada como se hizo en la Figura 1 para las emulsiones de betún asfáltico. La concentración de tensioactivo a la que se alcanza el valor máximo de ζ es la necesaria para formar una monocapa de BDTA en la interfase. Lane y Ottewill demuestran que, en el caso del betún asfáltico, cuanto más baja es la concentración micelar crítica (cmc) del tensioactivo, antes se llega a la saturación de la superficie, y ésta siempre tiene lugar a una concentración menor que la cmc. Después del máximo de ζ, y según los datos bibliográficos, en unos casos aparece una zona de “plateau” con ζ constante o un máximo al que sucede una bajada de ζ. Según Riddick (3), se produce un máximo cuando el medio de dispersión está concentrado y una zona de “plateau” cuando está diluido. En la zona de “plateau” se supone que se adsorben cantidades equivalentes de cationes del tensioactivo y de contraiones dentro de la partícula cinética. En la zona de descenso, el aumento de la fuerza iónica en el medio de dispersión concentrado origina una contracción de la capa doble, con disminución de ζ. El valor máximo que alcanza el potencial electrocinético depende de la capacidad de adsorción apolar de la superficie neutralizada del coloide, además de la dependencia obvia de las características iónicas del medio dispersante.

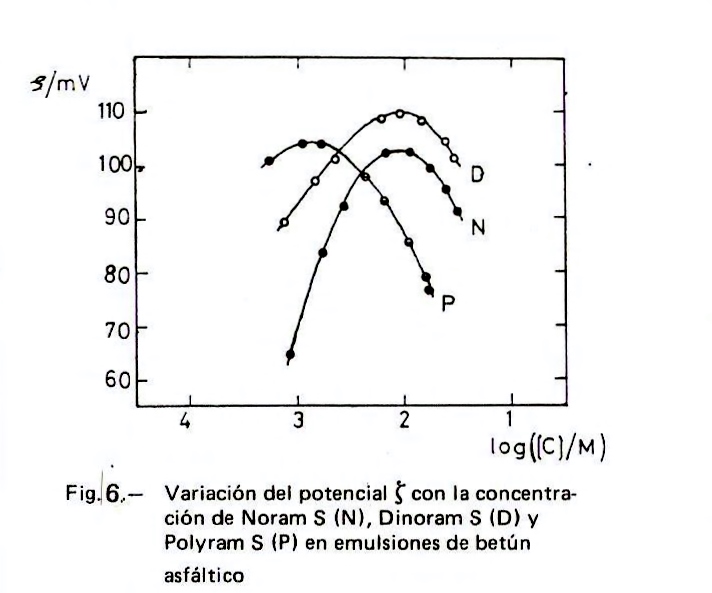
Para la fabricación industrial de emulsiones catiónicas de betún asfáltico se suelen emplear tensioactivos con un radical hidrocarbonado procedente de ácido graso con grupos polares –NH2 o –N(CH3)3. Hulshof (11) evalúa la variación del potencial zeta de las emulsiones de betún asfáltico con el pH y encuentra un máximo de ζ para un pH de 3 a 4 en el caso de cantidades de 0,25 y 0,35 por ciento de N-alquilpropilén 1,3 diamina con radical procedente de sebo (estas son cantidades normales en la producción industrial de las emulsiones bituminosas) y un máximo de ζ en un intervalo de 4 a 6 para una sal de amonio cuaternario. Uno de los autores del presente artículo (12) determinó la variación del potencial zeta con la concentración de los clorhidratos de alquilamina (representado en la Figura 6 con la letra N), de alquilpropiléndiamina (representado con la letra D) y de alquilpolipropilénpoliamina (representado con la letra P) con radical procedente de sebo en emulsiones de betún asfáltico. Como puede verse en la figura, el clorhidrato de poliamina satura la superficie del betún a menores concentraciones que los otros dos, como corresponde a su mayor carga positiva por molécula, lo que supone una mayor repulsión entre los extremos polares: cada molécula ocupa una mayor superficie. Los valores de ζ se midieron hasta donde lo permitía el método. No obstante, queda patente la disminución de ζ al aumentar la concentración de tensioactivo más allá del máximo, produciéndose una acción coagulante que está relacionada con la contracción de la capa doble; este efecto se hace mayor cuando hay sales disueltas en el medio dispersante (como suele ocurrir en las fabricaciones industriales), ya que aumenta la fuerza iónica.
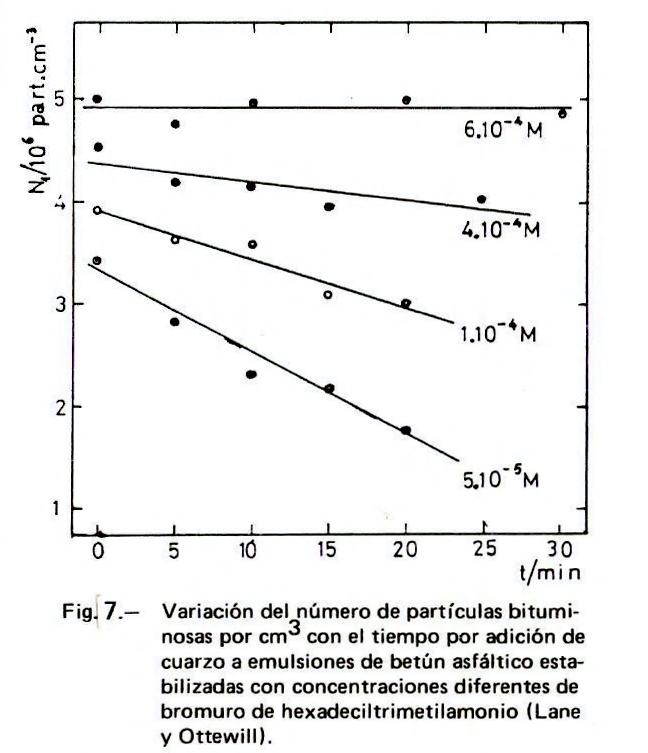
Para establecer el mecanismo de rotura de las emulsiones bituminosas catiónicas con los áridos silíceos se pueden enfrentar partículas de ambas clases. Así Lane y Ottewill miden la rotura de una emulsión bituminosa (tamaño de partícula de 1 a 5 µm) estabilizada con bromuro de hexametiltrimetilamonio (BHTA) en presencia de partículas de cuarzo de diámetros comprendidos entre 152 y 178 µm. Como se muestra en la Figura 7, por debajo de una concentración de 5.10-4M, que es la concentración a la que ζ alcanza el valor máximo, las partículas bituminosas heterocoagulan con las partículas de cuarzo a una velocidad que aumenta al disminuir la concentración del tensioactivo. Cuando se alcanza el máximo valor de ζ, la heterocoagulación no tiene lugar, debido a que la adsorción del tensioactivo (en exceso a una monocapa) sobre la superficie del cuarzo es más rápida que la adsorción de las partículas de betún, encontrándose éstas con partículas de cuarzo con carga positiva. Esta interpretación tiene validez, lógicamente, sólo para el tiempo del experimento y para la superficie específica del cuarzo empleado. Los resultados indican que la zona de ζ máximo es crítica. Si se requiere que la emulsión tenga una rotura rápida no debe sobrepasarse esta concentración.
Potencial zeta en dispersiones aniónicas y en áridos calizos
En las dispersiones aniónicas, la estructura de la capa doble se considera análoga a la de las dispersiones catiónicas, pero la superficie y la partícula cinética han de presentar una carga neta negativa, y la capa difusa una carga neta positiva de contraiones que compense la carga de la capa de Stern.
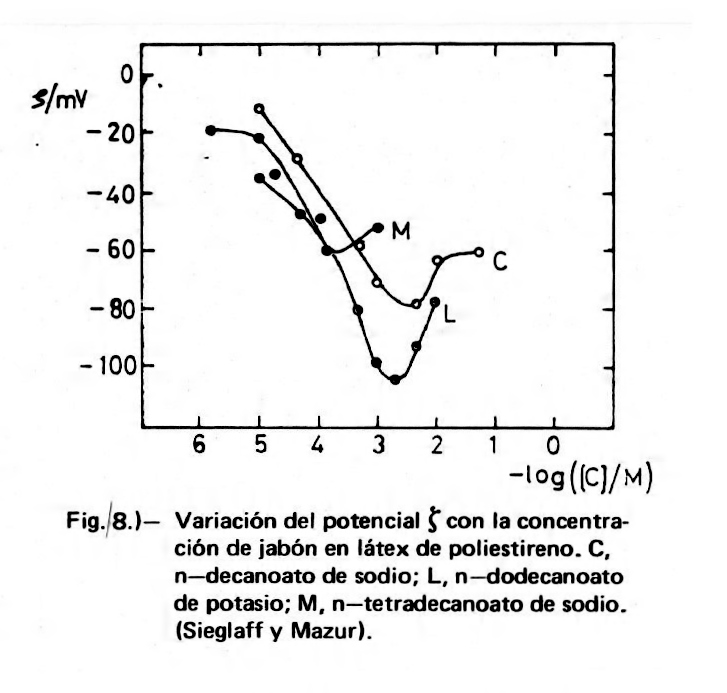
En la Figura 8 se compilan los datos de Sieglaff y Mazur (13) sobre la variación del potencial zeta en un látex de poliestireno monodisperso (1,7 µm de diámetro de partícula) con la concentración de tensioactivos aniónicos (extrajeron el emulsionante original mediante diálisis del látex). Como puede verse, los tres jabones ensayados, n-decanoato de sodio (caprato, C), n-dodecanoato de potasio (laurato, L) y n-tetradecanoato de sodio (miristato, M), dan máximos diferentes de potencial zeta, lo que indica la diferencia de adsorción de los iones del tensioactivo sobre la partícula de látex. Esta adsorción ha de realizarse por interacciones de van der Waals (asociación hidrofóbica) entre el radical apolar del tensioactivo y las cadenas apolares del poliestireno. Los mismos autores obtienen también diferencias en el valor máximo de ζ entre jabones sódicos y potásicos, dando estos últimos un potencial zeta ligeramente mayor.
En las emulsiones bituminosas aniónicas, las curvas de variación del potencial zeta con la concentración del tensioactivo serán similares a las mostradas para el látex. Como con algunos betunes asfálticos se pueden fabricar emulsiones estables a pH mayor de 9,5 sin adición de tensioactivo, posiblemente debido a la ionización de los ácidos nafténicos del betún, no se dará inversión de carga en las emulsiones bituminosas aniónicas.
Somasundaran y Agar (14) estudian el mecanismo de generación de carga en la superficie de una calcita mediante medidas del potencial de flujo. En la Figura 9 se exponen algunos de sus resultados relativos a la variación del potencial zeta con el tiempo y a pH diferentes; estos datos se muestran de acuerdo con el mecanismo de generación de carga que se expone a continuación. Los iones superficiales –Ca+ y –CO3– de la calcita pueden sufrir hidrólisis acompañada por la difusión de H+ u OH– a través de la capa doble hacia dentro o hacia fuera de la interfase. Así, en una disolución ácida, la hidrólisis de los iones carbonato superficiales -CO3– + H2O = -CO3H + OH– hace al mineral más positivo y a la disolución más alcalina. Análogamente, en el intervalo alcalino, la hidrólisis de los iones calcio es la responsable de los cambios de ζ observados: –Ca+ + H2O = -CaOH + H+. En este caso, la superficie se hace menos positiva y el pH de la disolución disminuye con el tiempo.
La velocidad de rotura de las emulsiones bituminosas aniónicas al enfrentarse con áridos minerales calizos depende, como en las catiónicas, de la magnitud del potencial zeta con respecto a la posición del máximo y de las características superficiales del material granular. Como ocurre con el cuarzo o la sílice coloidal, el desarrollo de la carga superficial de la calcita depende, no sólo de las características del medio acuoso, sino de la naturaleza cristalina del propio material. Así, Riddick (3) da como valor del potencial zeta de un carbonato cálcico finamente dividido -17±3 mV en un intervalo de concentraciones de 0,3 a 36 por ciento, siendo el pH del medio dispersante, agua, de 7,2 a 8,2. Considerando que el mecanismo de rotura, al igual que las emulsiones catiónicas con minerales silíceos, procede de las atracciones coulombianas entre las partículas cargadas negativamente de la emulsión y las cargas positivas superficiales del mineral (que siempre existen por haber –Ca+) la velocidad de rotura dependerá también de la magnitud de esta carga positiva, y se sabe que será mayor en medio ácido. En la práctica, hay que tener en cuenta el carácter dinámico de las superficies calizas por el establecimiento del equilibrio de hidrólisis.

Información obtenible del potencial zeta para la fabricación y empleo de las emulsiones de betún asfáltico
La determinación del potencial electrocinético ζ se muestra como un dato útil en la evaluación de las características dinámicas tanto de la emulsión bituminosa como de las cualidades superficiales de los áridos minerales con los que ha de enfrentarse. Como los valores de ζ vienen determinados por la longitud y naturaleza de la cadena hidrofóbica y por el tamaño y la carga del grupo hidrofílico, su medida puede determinar la elección de los agentes emulsionantes y la formulación de las emulsiones. Asimismo, puede servir para el análisis de las emulsiones empleadas en la práctica.
Para formular una emulsión de rotura rápida es conveniente que el porcentaje de tensioactivo no sobrepase el valor máximo del potencial zeta. En cambio, para fabricar emulsiones de rotura media o lenta habría que sobrepasar dicho máximo o, mejor, determinar los emulsionantes y los porcentajes adecuados para los empleos que se requieran.
En los apartados anteriores se ha visto que existe un intervalo de pH de la emulsión que optimiza el valor de ζ. Este intervalo de pH está relacionado, sin duda, con el equilibrio de disociación del emulsionante y con la capacidad de adsorción de los iones del tensioactivo sobre la superficie de las partículas. Por lo tanto, hay que tener en cuenta el pH en la formulación de la emulsión, ya que puede conducir a valores distintos de ζ. Además, en las fabricaciones industriales de emulsiones, hay que notar que normalmente se trabaja con aguas que contienen cantidades variables de iones en disolución. Estos iones, por adsorberse en la partícula cinética, modifican tanto el valor de ζ como la posición del máximo, y la cuantía de las modificaciones depende de su carga iónica y de su concentración. En general, el máximo de ζ se obtiene con tanta menor concentración de tensioactivo cuanto mayor sea la fuerza iónica del medio. Los iones disueltos, como el exceso del propio emulsionante, actúan como floculantes de la emulsión.
En el hecho físico del enfrentamiento de las emulsiones bituminosas con los áridos calizos o silíceos en el trabajo de construcción de carreteras, la generación de cargas superficiales en los áridos depende no sólo de su humedad, sino de la velocidad con que se manifieste la influencia del medio dispersante de la emulsión sobre su superficie. Si se alcanza el equilibrio iónico con rapidez suficiente, se darán las situaciones que se exponen a continuación. Una emulsión catiónica, con pH ácido, disminuirá el valor de -ζ en un árido silíceo. Si se requiere una velocidad de rotura máxima, habrá que operar a valores de pH para los que la diferencia entre el potencial +ζ de la emulsión y el -ζ del árido sea máxima. Las emulsiones aniónicas, con pH básico, aumentarán el valor de -ζ de los áridos silíceos, con lo que se acentuarán las repulsiones coulombianas entre la superficie del sólido y las partículas de la emulsión. En el trabajo con emulsiones catiónicas y áridos calizos interesa que el pH sea lo más alto posible dentro del intervalo óptimo si se requiere rotura rápida, ya que así aumentará el valor de -ζ. Con emulsiones aniónicas y áridos calizos, la velocidad de rotura aumentará cuando el pH de la emulsión esté en los valores bajos del intervalo óptimo.
Hasta aquí se ha intentado hacer una traslación cualitativa de los datos bibliográficos al problema íntimo del mecanismo molecular de la rotura de la rotura de las emulsiones bituminosas frente a los áridos minerales. Es evidente que existe una documentación experimental escasa, pero si se quieren efectuar determinaciones cuantitativas, la medida del potencial electrocinético se muestra como una herramienta interesante.
BIBLIOGRAFÍA
1.- W.D. Harkins; “The physical chemistry of surface films”, Reinhold Pub. Corp., N.Y.,1952
2.- D. Stigter; J. Phys. Chem., 68, 3603 (1964); 78, 2480 (1974); 79, 1008 (1975). J. Colloid Interface Sci., 23, 379 (1967); 47, 473 (1974).
3.- T.M. Riddick; “Control of colloid stability through zeta potential”, Zeta-meter Inc., N.Y.,1968.
4.- H.C. Li y P.L. De Bruyn; Surface Sci., 5, 203 (1966).
5.- B. Dobiá; Colloid and Polymer Sci., 256, 203 (1978).
6.- R.H. Ottewill, M.C. Rastogi y A. Watanabe; Trans Faraday Soc., 56, 854, 866, 880 (1960).
7.- J.B. Kayes; J. Colloid Interface Sci., 56, 426 (1976)-
8.- J.P. Friend y R.J. Hunter; J. Colloid Interface Sci., 37, 548 (1971).
9.- B.H. Bijsterbosch; J. Colloid Interface Sci., 47, 186 (1974).
10.- A.R. Lane y R.H. Ottewill; Theory Pract. Emulsion Technol., Proc. Symp., Academic, London, 1976.
11.- W.T. Hulshof; Bull. Liaison Labo. P. Et Ch., 94, 175 (1978).
12.- C. Santos; Tesis doctoral. Universidad de Alicante, 1977.
13.- C.L. Sieglaff y J. Mazur; J. Colloid Interface Sci., 15, 437 (1960); 17, 66 (1962).
14.- P. Somasundaran y G.E. Agar; J. Colloid interface Sci., 24, 433 (1967).
